- Introduction
- PepQuery
- Installation
- System requirements
- Web application
- Input data
- Parameters
- Datasets
- Result page
- Stand alone version
- Input data
- Parameters
- Output data
- Result visualization
- Multiple datasets
- Demo
- Indexing
- Immunopeptidomics
PepQuery
PepQuery is a universal targeted peptide search engine for identifying or validating known and novel peptides of interest in any local or publicly available mass spectrometry-based proteomics datasets. It's different from the spectrum-centric search engines, such as Mascot, MaxQuant and MS-GF+.
You can read the following papers to know more about how PepQuery version 1 works:
Wen, Bo, Xiaojing Wang, and Bing Zhang. "PepQuery enables fast, accurate, and convenient proteomic validation of novel genomic alterations." Genome research 29.3 (2019): 485-493.
Wen, Bo, Kai Li, Yun Zhang, and Bing Zhang. "Cancer neoantigen prioritization through sensitive and reliable proteogenomics analysis." Nature communications 11. 1 (2020): 1-14.
We recently developed PepQuery2 which leverages a new MS/MS indexing approach and cloud storage to enable ultrafast, targeted identification of both novel and known peptides. The standalone version of PepQuery2 allows users to search more than one billion MS/MS data indexed in the PepQueryDB from any computers with internet access. It also supports direct analysis of user provided MS/MS data, any public datasets in PRIDE, MassIVE, jPOSTrepo and iProX, or Universal Spectrum Identifiers (USIs) from ProteomeXchange. Meanwhile, we have extended the web version to include public proteomics datasets from all flagship CPTAC studies, leading to a total of 48 datasets.
You can read the following papers to know more about how PepQuery version 2 works:
Wen, Bo, and Bing Zhang. "PepQuery2 democratizes public MS proteomics data for rapid peptide searching." Nature Communications 14.1 (2023): 2213.
Installation
PepQuery is available as a standalone application as well as a PepQuery server (PepQuery Web). In order to run the standalone version of PepQuery, you must have Java 1.8 or newer installed. To check your java version please open your terminal application and run the following command:
java -version
Please go to the Download page to download the standalone version of PepQuery. It's written by Java and is platform independent. This package is a tar.gz file. After you download it, please uncompress it using the following command line and you will find a jar file in the package folder. The whole installation process typically only takes one or two minutes.
tar xvzf pepquery-2.0.2.tar.gz
If you want to take a Variant Call Format (VCF), Browser Extensible Data (BED) or Gene Transfer Format (GTF) file as input, you will need to install R and the R package PGA. You need to prepare annotation data according to the user's manual of PGA before you start to run PepQuery with taking VCF, BED or GTF file as input. PepQuery uses PGA to translate the events in the VCF, BED or GTF file to protein sequences. If you want to know more about PGA, please read the paper of PGA (doi: 10.1186/s12859-016-1133-3). If you only want to take a peptide, protein or DNA sequence as input, you don't need to install R and PGA.
System requirements
For PepQuery Web version, there is no specific need for the system. It only requires a web browser with internet connection.
For PepQuery standalone version, we recommend a computer with 8 GB of memory and 4 CPUs. Windows, Mac OS and Linux systems are supported.
The latest version of PepQuery is 2.0.2. We have tested this on different datasets.
Web application
You can use PepQuery through PepQuery Web. Using PepQuery Web, you don't need to prepare the MS/MS data and protein reference database. Of course, you also don't need to install the PepQuery software in your computer. All you need to provide is the target peptide/protein/DNA sequence or protein ID which you want to identify. Please note the searching using the web server could be reproduced using stand alone version of PepQuery. The parameter section of the result page contains the command line used for a searching. In addition, please note the web server only allows to submit one job at a time to make full use of the computer resource to speed up the search. When a job is running, users have to wait the job to be finished to view the main page or submit a new job.
Input data
Currently, the web interface accepts taking peptide, protein, DNA sequences or protein ID as input. For each search, only one sequence is supported. If you have multiple sequences, pelase do multiple searches or use the standalone version. The standalone version also supports to search all the indexed MS/MS datasets available in the web server in any local computer with internet connection.
Parameters
There are only a few parameters you need to set. Below is a screenshot of the web inferface parameters:
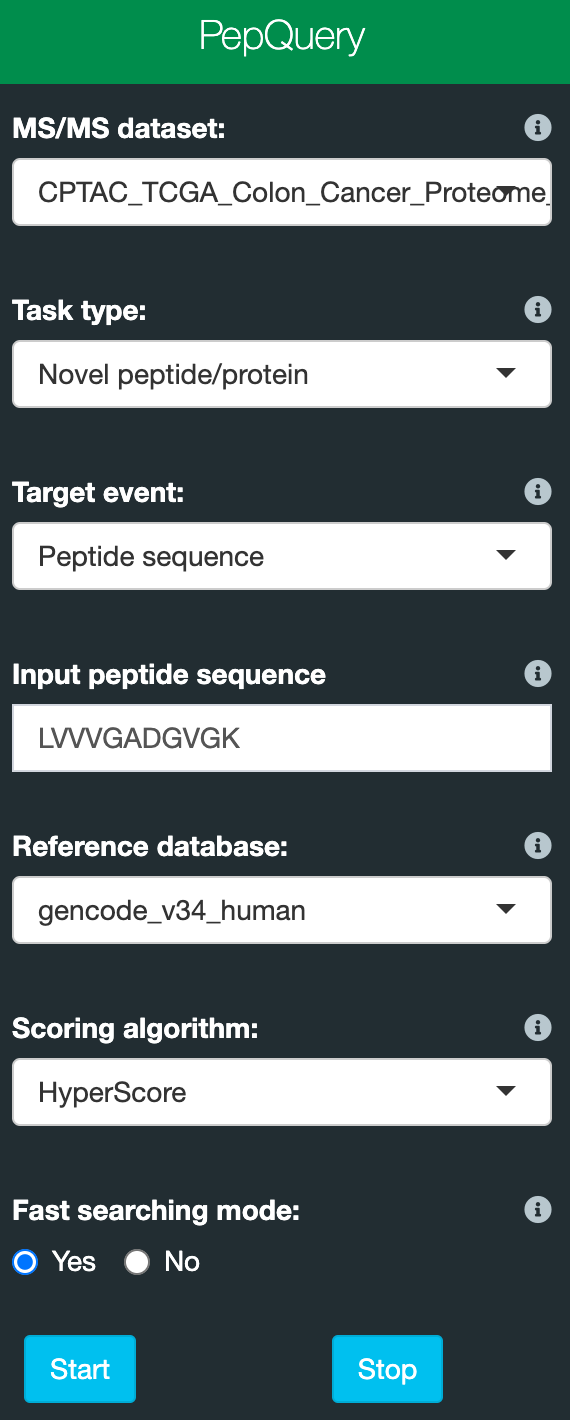
Please find the details about the parameters below:
- MS/MS dataset: select one MS/MS dataset which you want to search. Detailed information about the datasets can be found at the homepage of the web server: web server
- Task type: novel or known peptide/protein identification.
- Target event: select the sequence class you provide. For the web interface, peptide, protein and DNA sequences are supported for novel peptide/protein identification. Peptide sequence and protein ID are supported for known peptide/protein identification.
- Input peptide/protein/DNA sequence or a protein ID: a peptide/protein/DNA sequnce or a protein ID depends on your selection for target event. For one search, only one sequence is accepted.
- Reference database: a protein reference database;
- Scoring algorithm: peptide spectrum scoring algorithm: Hyperscore and MVH are available. Default is Hyperscore. We recommend to use Hyperscore.
- Fast searching mode: choose to use the fast mode for searching or not. In fast mode, only one better match from reference peptide-based competitive filtering steps will be returned. For most applications, 'Yes' should work fine. A peptide identified or not is not affected by this setting.
For each dataset in this web application, we have selected a set of optimized MS/MS searching parameters so that users don't need to set these parameters by theirself. In the result page, you can find the detailed MS/MS searching parameters for the selected MS/MS dataset. If you want to change any of the parameter settings for a dataset, you could use the standalone version.
Datasets
Detailed information about the datasets can be found at the homepage of the web server: web server
Result page
Through the result web page of a search, you can find the detailed identification result about your input sequence. The result page is divided into four panels:
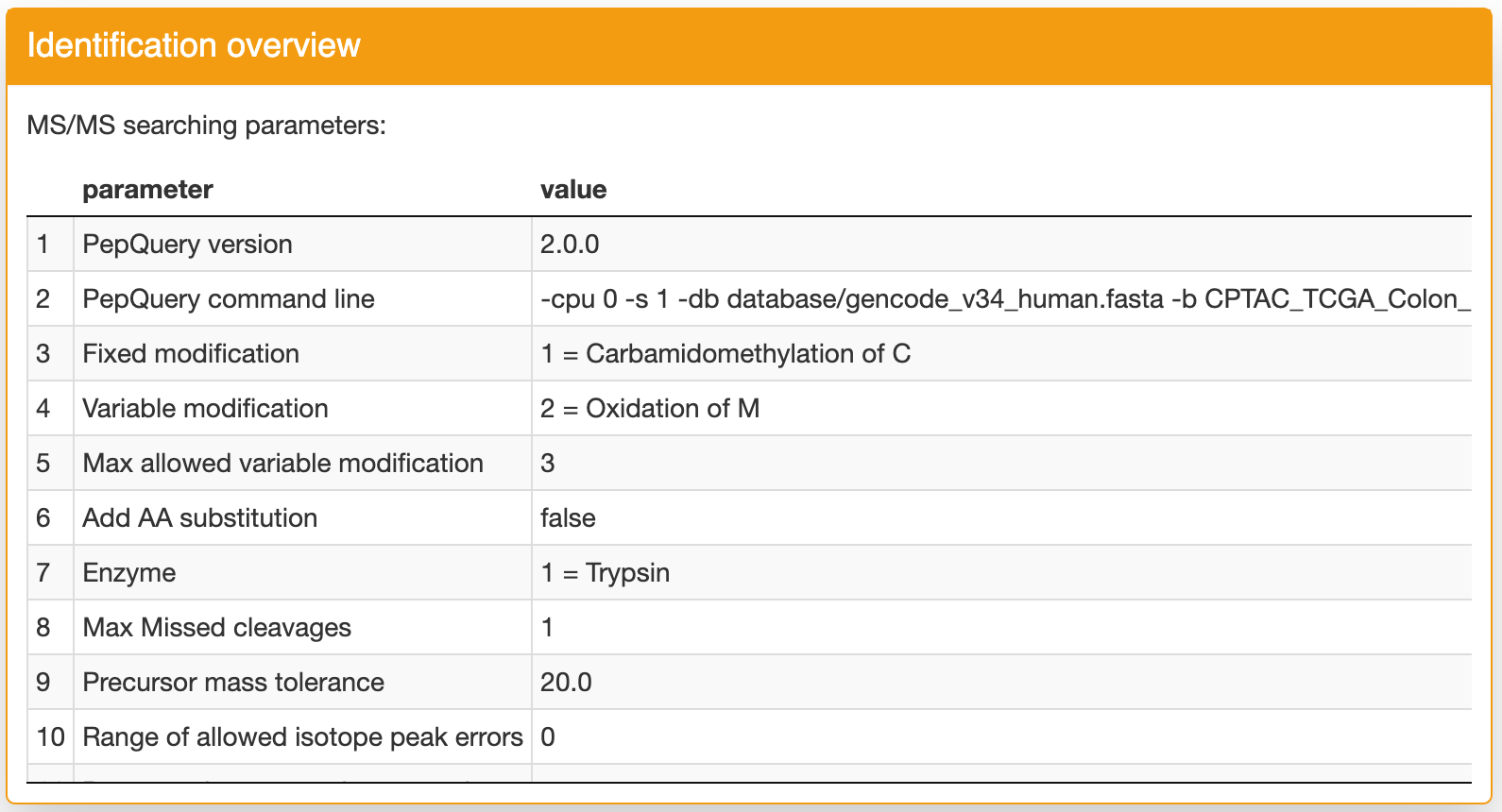
Identification overview: the first panel contains the identification parameters for the selected dataset:
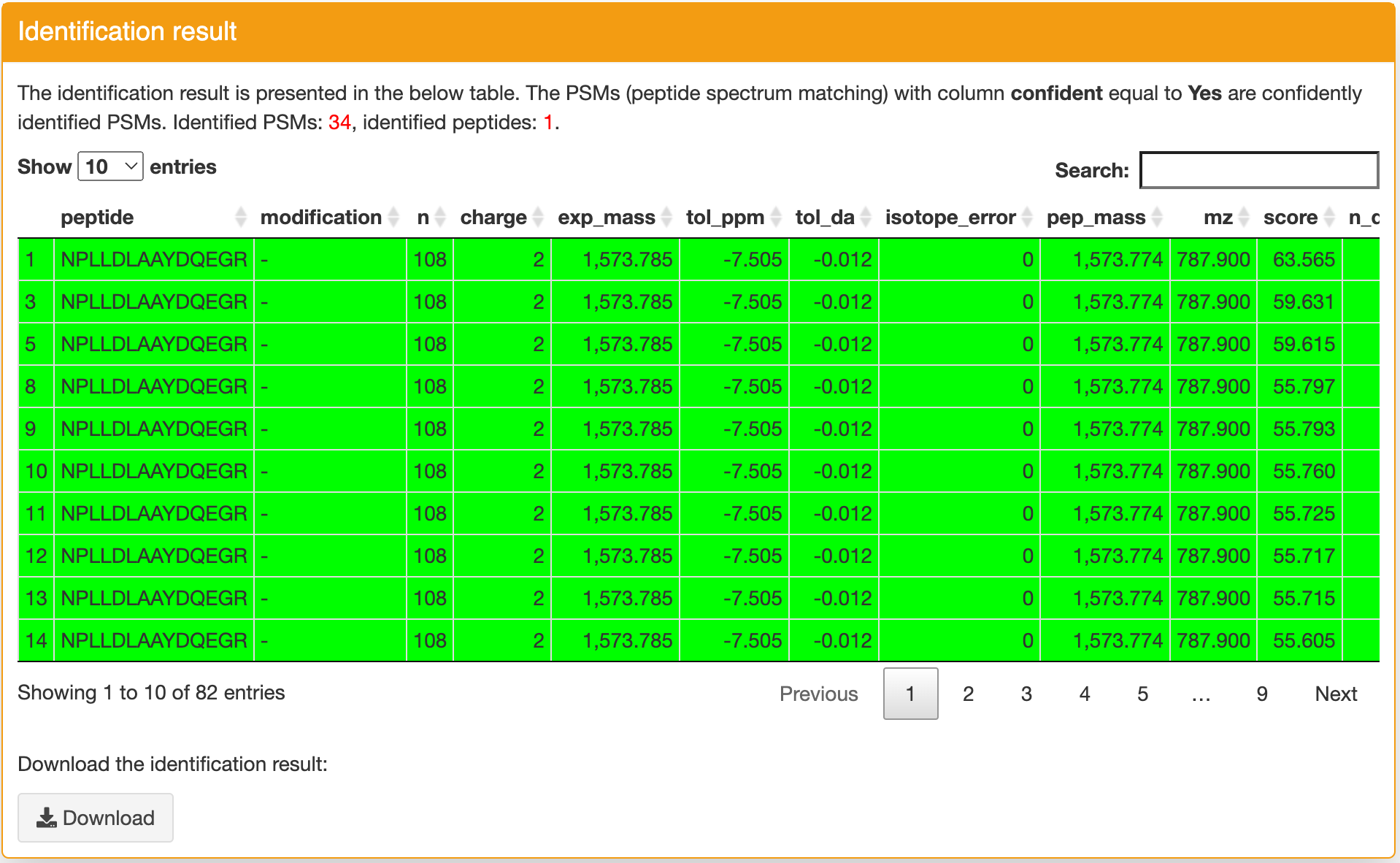
Identification result: this panel contains the detailed identification result for input sequence. If a row is green, it indicates this identification is confident (column confident == Yes). You can click a row then you can find the spectrum annotation figure in the "Spectrum annotation" panel. This figure can help you further evaluate the quality of the peptide spectrum matching. If you want ot download the identification table, you can click the "Download" button in the bottom of this panel. Using the "Search" function, you can quickly filter the rows you want. Please note, if there is any match passed the step 3 filtering, only the PSMs passed this filtering will be shown in the table to simplify the result. If there is no match passed the step 3 filering, all matches will be shown in the table. If there is no any match found, the detailed output log information will be shown.
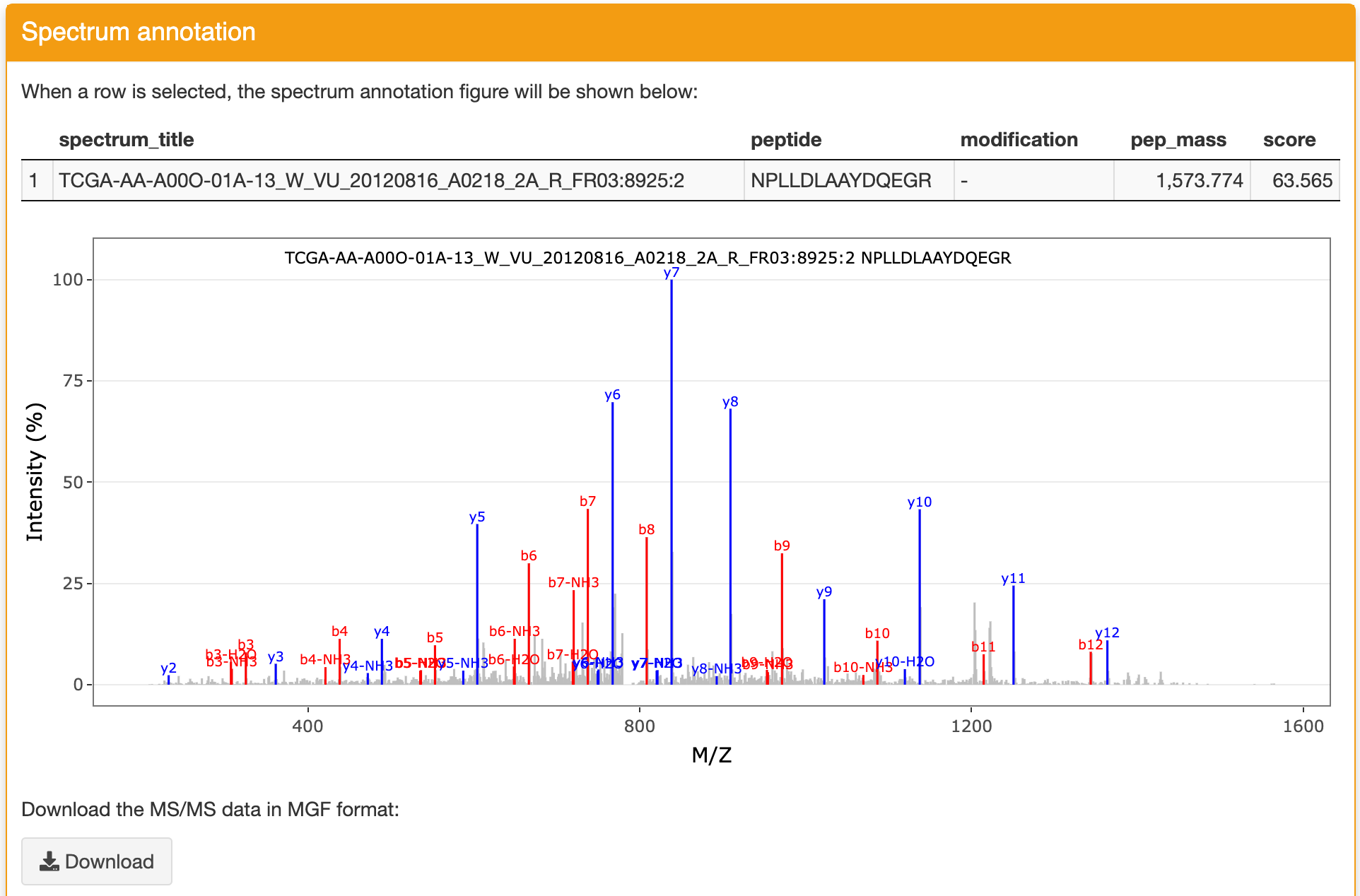
Spectrum annotation: this panel displays the spectrum annotation figure of the identification you selected in the Identification result panel. The matched ions are displayed with different colors. The figure can be zoomed in and zoomed out. You can download the figure and the MS/MS data refered to this identification through the functions in this panel.
The best matched peptide from the reference database for the selected spectrum is also listed in this panel as shown below:
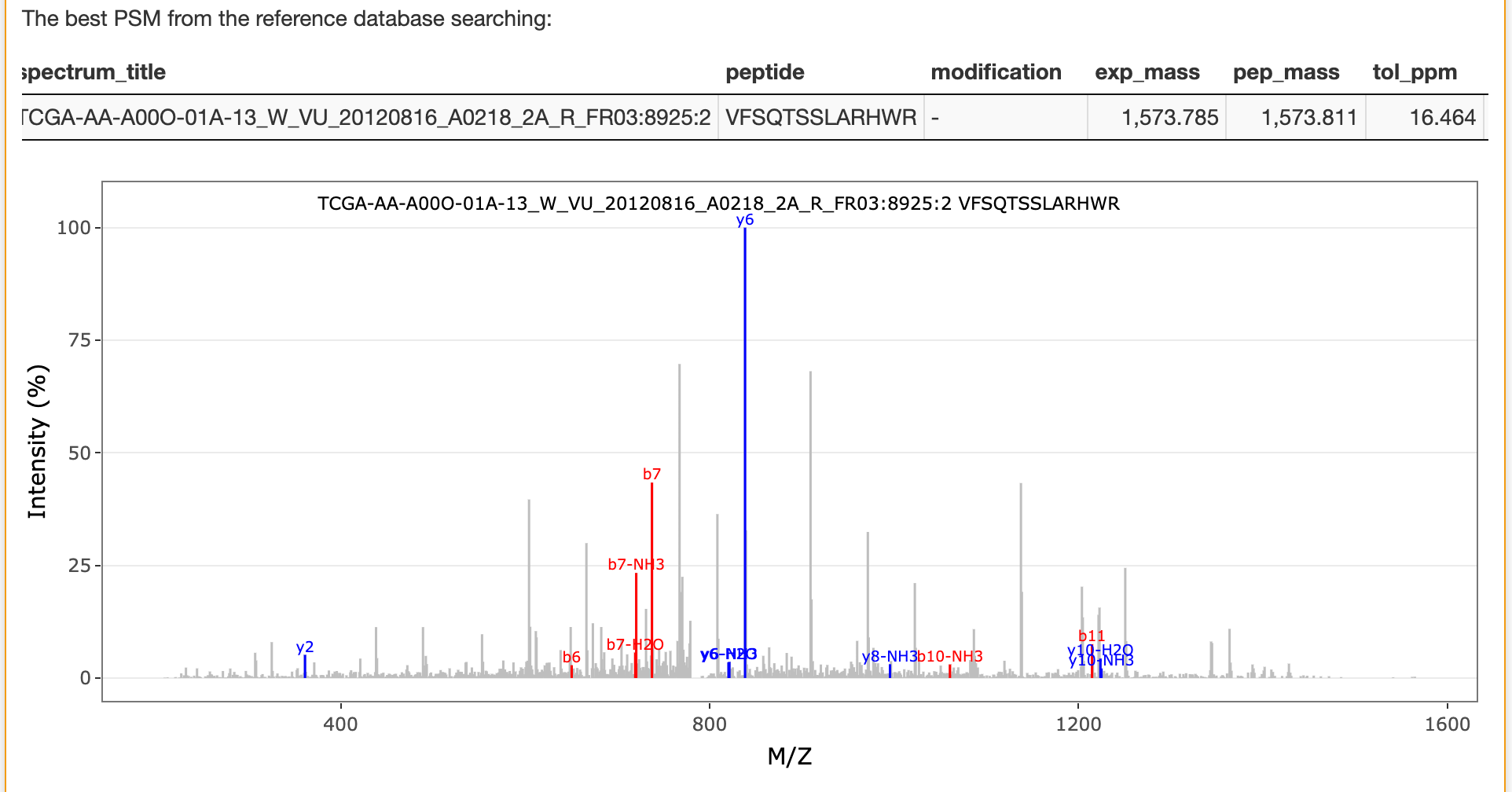
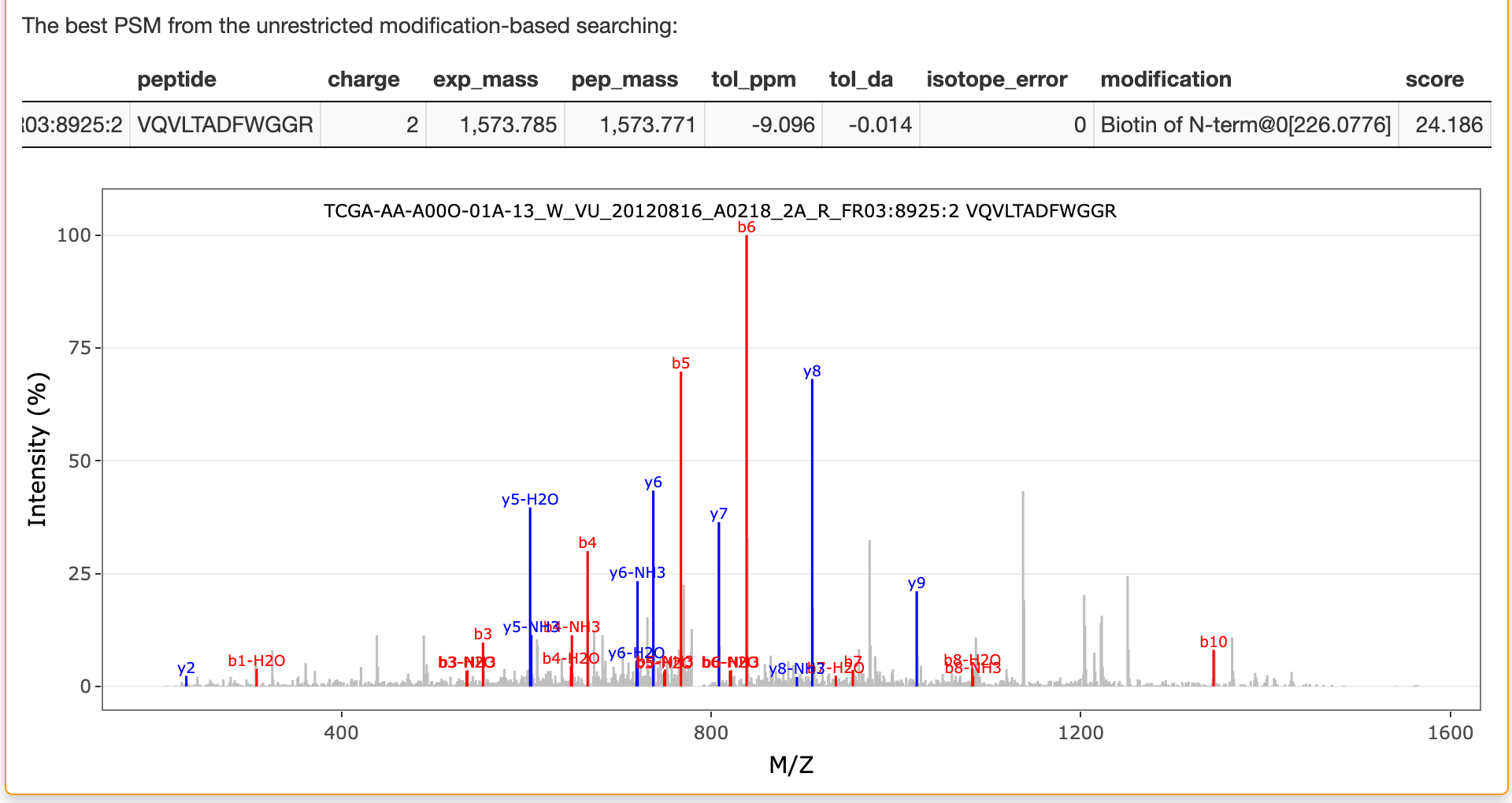
If unrestricted modification searching is selected, the best matched peptide from the reference database for the selected spectrum in this searching is also listed in this panel as shown below:
Sample information: you can find the sample information for the selected row in the "Identification result" panel.

Standalone version
Input data
As for the standalone version of PepQuery, usually you need to provide MS/MS data in MGF format, a reference protein database in FASTA format and a peptide, protein, DNA sequence or a pair of peptide and spectrum ID. However, if you want to take a VCF, BED or GTF as input, you will need to provide a folder which includes annotation data. This annotation data will be used to translate the input events into protein sequences.
Parameters
Please find the command line parameters of PepQuery below:
- -i: The value for this parameter could be:
(1) a single peptide sequence;
(2) multiple peptide sequences separated by ',';
(3) a file contains peptide sequence(s). If it is a file contains peptide sequence(s), each row is a single peptide sequence and there is no header in the file;
(4) a file contains peptide sequence and spectrum ID and they are separated by tab '\t'. There is no header in the file. This is used when perform PSM level validation;
(5) a protein sequence. This is used when perform novel protein identification;
(6) a protein ID. This is used for targeted known protein identification;
(7) a DNA sequence;
(8) a VCF file;
(9) a BED file;
(10) a GTF file. - -t: Input type for parameter -i: peptide (or pep) when the setting for -i belongs to (1)-(4), protein (or pro) when the setting for -i belongs to (5)-(6), DNA (or dna), VCF (or vcf), BED (or bed), GTF or (gtf). The default is peptide.
- -s: Task type: 1 => novel peptide/protein validation (default), 2 => known peptide/protein validation. Default is 1.
- -ms: MS/MS data used for identification. MGF, mzML, mzXML, mgf.gz, mzML.gz, mzXML.gz and Thermo raw formats are supported.
MGF/mzML/mzXML format files can be converted from raw files using msconvert before running PepQuery:
For example, from raw MS/MS data to MGF data:
msconvert --filter "peakPicking true 1-2" --mgf *.raw
From mzML format data to MGF data:
msconvert --filter "peakPicking true 1-2" --mgf *.mzML
To show all the indexed MS/MS datasets in PepQueryDB:
java -jar pepquery-2.0.2.jar -b show
To show all the supported modifications in PepQuery:
java -jar pepquery-2.0.2.jar -printPTM
To show all predefined parameter settings in PepQuery:
java -jar pepquery-2.0.2.jar -p show
Output data
PepQuery outputs results as several tab-delimited text files.
psm_rank.txt: this is the mainly result file of PepQuery. This file includes the detailed identification results for the input target peptide. We recommend users to filter the identification result based on the column "confident" in this file with confident equal to "Yes". If a PSM with "Yes" in column "confident", it means this identification passed all the validation steps in PepQuery.
| Column | Description |
|---|---|
| peptide | A target peptide sequence. |
| modification | Modification information of the target peptide. For example, "Deamidation of N@6[0.9840]". "-" means there is no modification. |
| n | The number of candidate spectra for the target peptide. |
| spectrum_title | Sepctrum title of the matched spectrum. |
| charge | The charge state of the precursor ion. |
| exp_mass | The experimental mass of the matched spectrum. |
| tol_ppm | Mass error in ppm unit. |
| tol_da | Mass error in Da unit. |
| isotope_error | Isotope error. |
| pep_mass | The theoretical mass of the peptide. |
| mz | The mass-over-charge value of the precursor ion. |
| score | The score of the identification, where larger is better. |
| n_db | The number of better matched peptides from the reference protein database to the matched spectrum. "-1" means that the competitive filtering based on reference sequences (step 3) is not performed for this match because it fails in a previous validation step. "0" means that there is no better match found from the competitive filtering based on reference sequences (step 3). If n_db >= 1, it means there are "n_db" better matches found from the competitive filtering based on reference sequences step. |
| total_db | The total number of matched peptides from the reference protein database without regarding the score. |
| n_random | The number of better matched random peptides to the matched spectrum. |
| total_random | The total number of random peptides. |
| pvalue | The pvalue of the identification, where smaller is better. If the pvalue for a PSM is 100 in the file, it means the corresponding PSM is not confident and is not necessary to have p-value validation (step 4). This is because this PSM is found to be not confident based on the processing in step 2-3 showing in the Figure 2 of the PepQuery paper. |
| rank | The rank of the identification. |
| n_ptm | The number of better matched modification peptides when performing the unrestricted modification searching. "-1" means that there is no unrestricted modification searching done for this match because it fails in a previous validation step. "0" means that there is no better match found from the unrestricted modification searching. If n_ptm >= 1, it means there are n_ptm better matches found from the unrestricted modification searching step. |
| confident | Yes: the PSM is confident, No: the PSM is not confident. Users can directly use this to filter the identification results. |
| ref_delta_score | The delta score between the match to the target peptide and the "best match" from competitive filtering based on reference sequences (step 3). In fast model when '-fast' is set, the "best match" may not be real best match. |
| mod_delta_score | The delta score between the match to the target peptide and the "best match" from competitive filtering based on unrestricted post-translational modification searching. (step 5). In fast model when '-fast' is set, the "best match" may not be real best match.. |
psm_rank.mgf: this is an MGF file which contains all the MS/MS spectra in psm_rank.txt file. The users can extract the MS/MS spectra from this file if they want to do some downstream analysis.
ptm_detail.txt: this file contains the identifications of better matched modification peptides from the unrestricted modification searching. It's possible to be empty when there is no better match. Except containing all the columns in psm_rank.txt, it also contains the following columns:
| Column | Description |
|---|---|
| ptm_spectrum_title | Sepctrum title of the matched spectrum. |
| ptm_peptide | Modification peptide sequence. |
| ptm_charge | The charge state of the precursor ion. |
| ptm_exp_mass | The experimental mass of the matched spectrum. |
| ptm_pep_mass | The theoretical mass of the peptide. |
| ptm_tol_ppm | Mass error in ppm unit. |
| ptm_tol_da | Mass error in Da unit. |
| ptm_isotope_error | Isotope error. |
| ptm_modification | Modification information of the target peptide. For example, "Carbamidomethylation of C@2[57.0215];Methyl of C@2[14.0157]". |
| ptm_score | The score of the identification, where larger is better. |
ptm.txt: this file contains all the identifications from competitive filtering based on unrestricted post-translational modification searching. (step 5). All the modification peptides in ptm_detail.txt which are from the unrestricted modification searching are also included in this file.
| Column | Description |
|---|---|
| spectrum_title | Sepctrum title of the matched spectrum. |
| peptide | Modification peptide sequence. |
| charge | The charge state of the precursor ion. |
| exp_mass | The experimental mass of the matched spectrum. |
| pep_mass | The theoretical mass of the peptide. |
| tol_ppm | Mass error in ppm unit. |
| tol_da | Mass error in Da unit. |
| isotope_error | Isotope error. |
| modification | Modification information of the target peptide. For example, "Carbamidomethylation of C@2[57.0215];Methyl of C@2[14.0157]". |
| score | The score of the identification, where larger is better. |
detail.txt: this file contains all the identifications from competitive filtering based on reference sequences (step 3).
| Column | Description |
|---|---|
| spectrum_title | Sepctrum title of the matched spectrum. |
| peptide | peptide sequence. |
| modification | Modification information of the target peptide. For example, "Carbamidomethylation of C@2[57.0215]". |
| exp_mass | The experimental mass of the matched spectrum. |
| pep_mass | The theoretical mass of the peptide. |
| tol_ppm | Mass error in ppm unit. |
| tol_da | Mass error in Da unit. |
| isotope_error | Isotope error. |
| score | The score of the identification, where larger is better. |
psm_type.txt: this file contains PSM level validation result. This is only available when performing PSM level validation.
| Column | Description |
|---|---|
| peptide | peptide sequence. |
| spectrum_title | Sepctrum title of the matched spectrum. |
| type | The type of PSM validation result: no_spectrum_match, low_score, high_p_value, better_ref_pep, better_ref_pep_with_mod, confident and invalid_peptide. |
parameter.txt: this file contains the parameters used for PepQuery analysis.
Result visualization
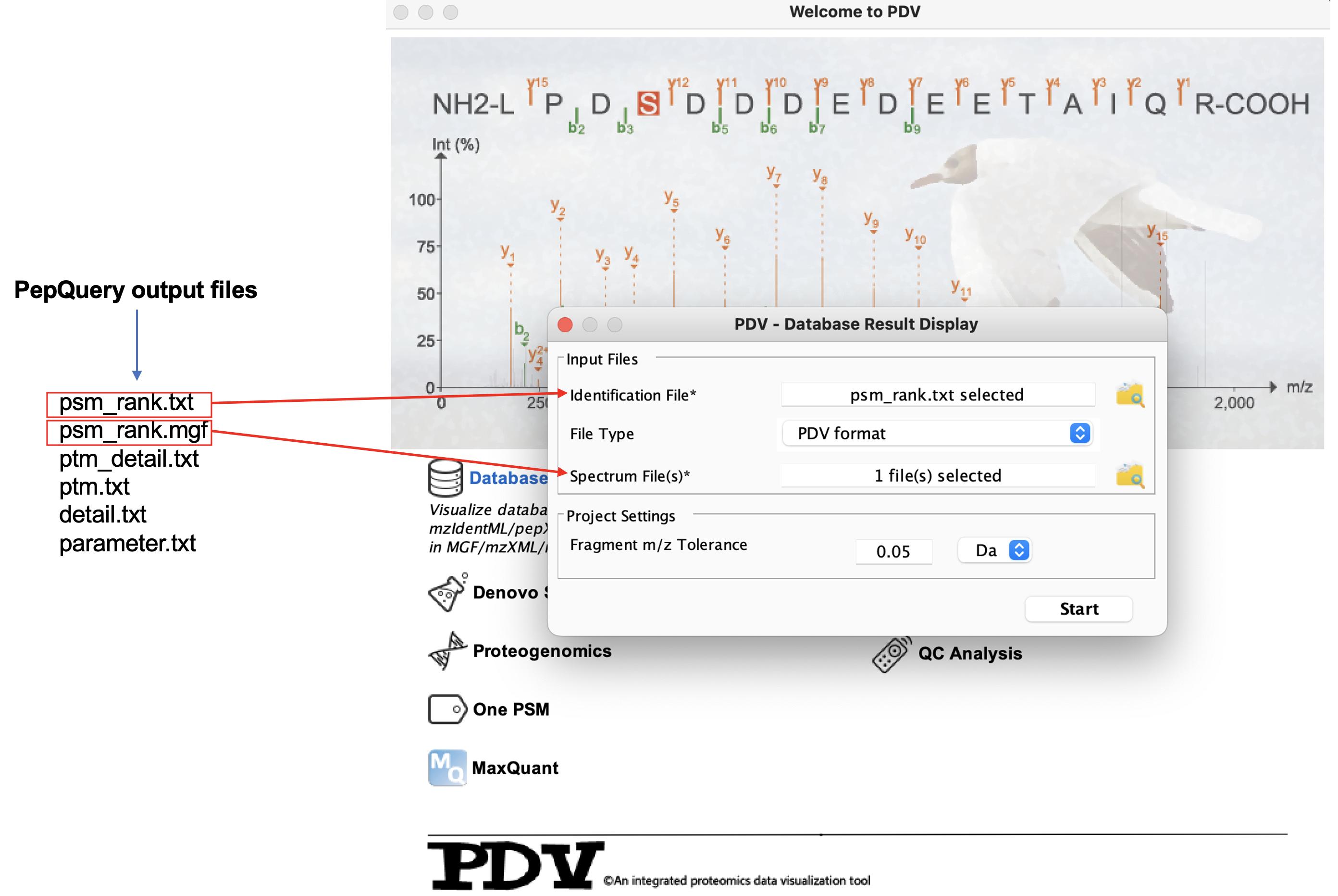
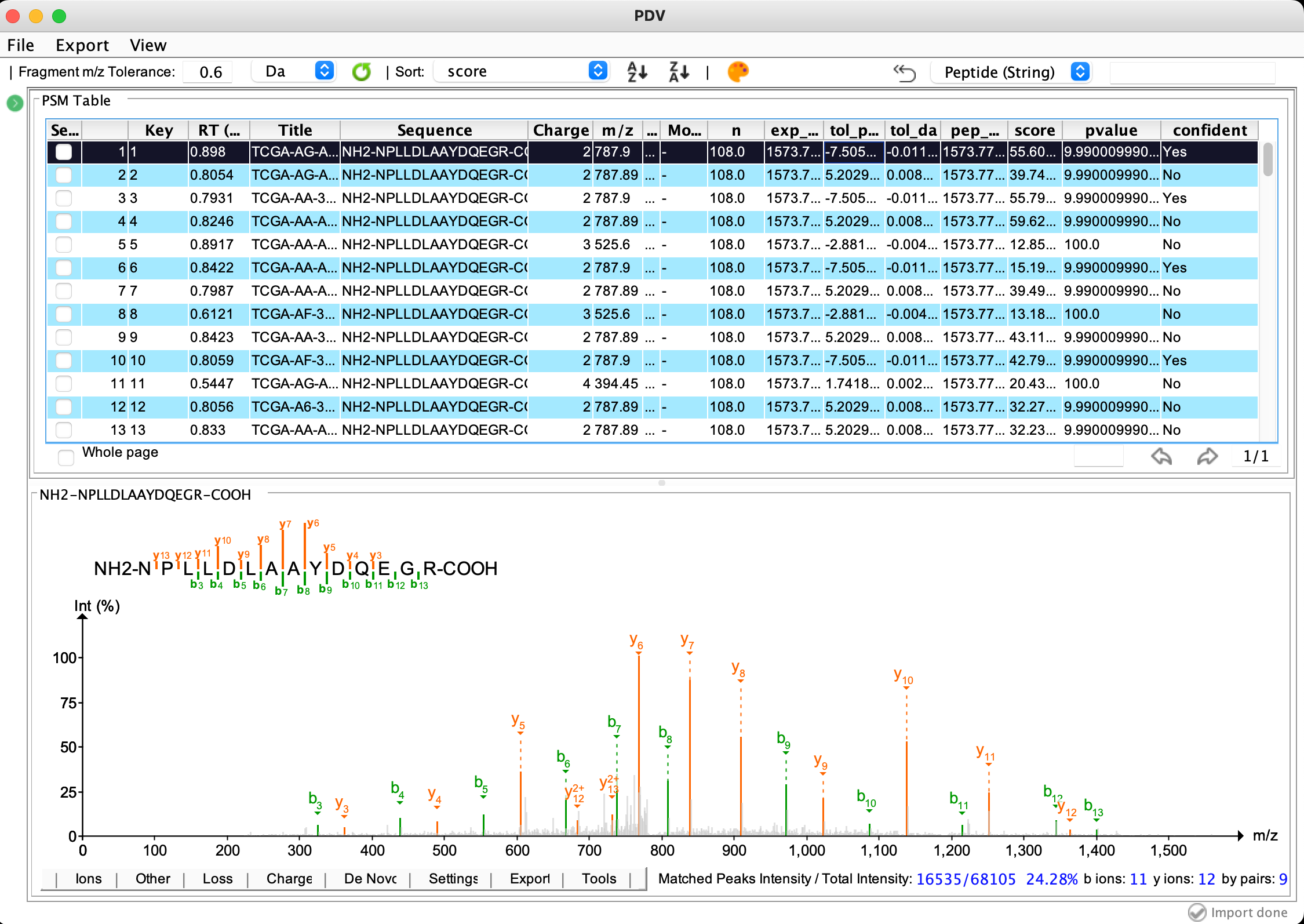
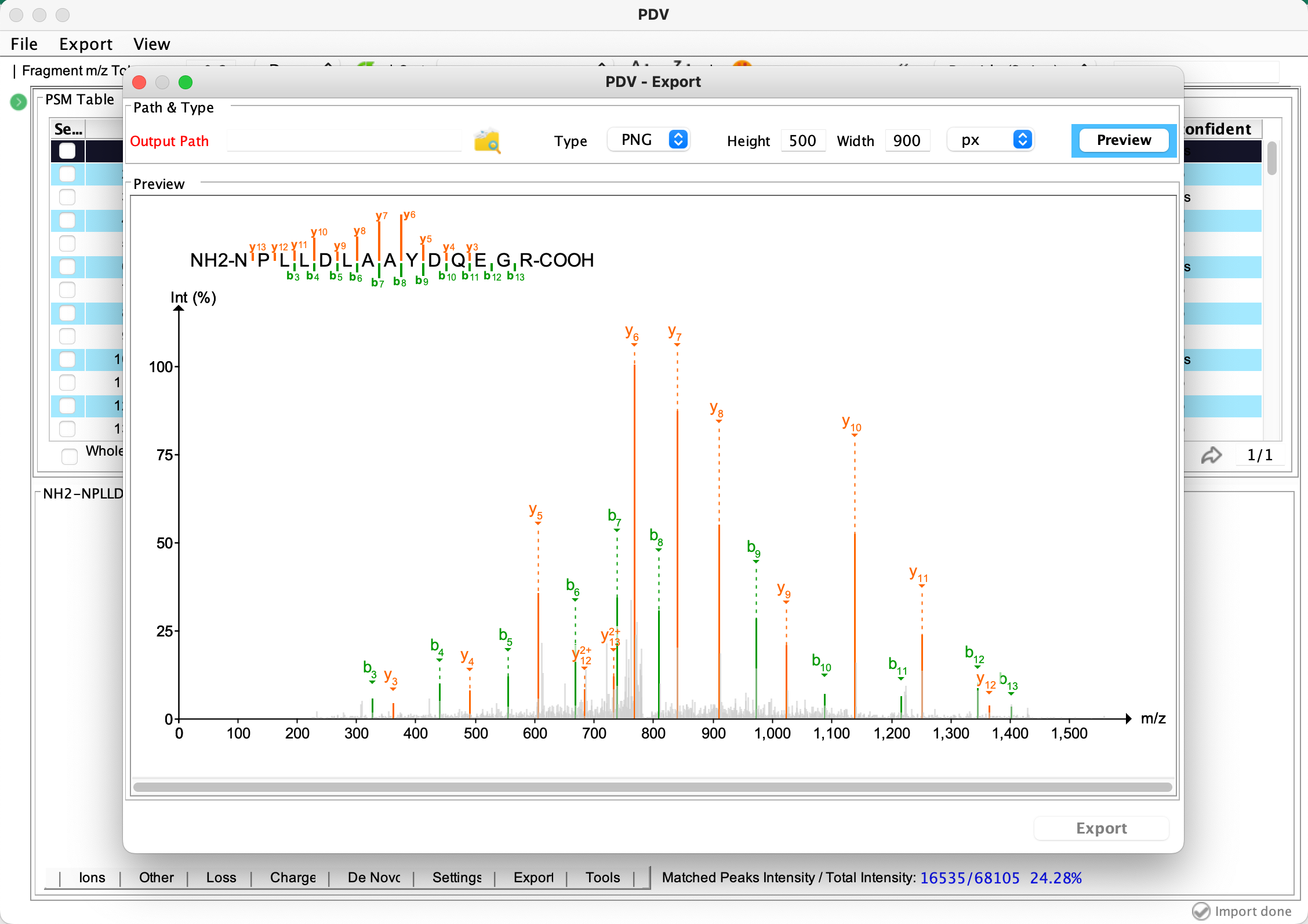
The result files of PepQuery can be imported to PDV which is developed by our lab to visualize. Currently, the file psm_rank.txt, ptm.txt or detail.txt along with the psm_rank.mgf can be imported into PDV for visualization. Through PDV, you can check the spectrum annotation figure for each identification one by one and you can also export high-quality annotation figure in different formats such as png and pdf. This function can help you further evaluate the quality of the identification manually.
Below is how to load the PepQuery identification result (psm_rank.txt and psm_rank.mgf) into PDV for visualization and how it looks like:
Load the data into PDV:
The main visualization panel:
Export a selected annotated spectrum in PNG or PDF format:
Searching multiple datasets in PepQueryDB
It's very easy to search multiple MS/MS datasets available in PepQueryDB in a single analysis. Below is an example:
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b CPTAC_LUAD_Discovery_Study_Proteome_PDC000153,CPTAC_PDA_Discovery_Study_Proteome_PDC000270,CPTAC_UCEC_Discovery_Study_Proteome_PDC000125 -db gencode:human -hc -o pepquery_kras_g12d/ -i LVVVGADGVGK
Demo
1. Search a novel peptide against a single MS/MS dataset from PepQueryDB which contains 48 indexed proteomics datasets. All the proteomics data in PepQueryDB can be directly accessed through PepQuery standalone version.
The following command line searchs the KRAS G12D variant peptide "LVVVGADGVGK" in the CPTAC LUAD global proteome dataset (Use command line "java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b show" to show all available datasets in PepQueryDB or go to PepQuery Web version to look at the available dataset information). The reference database used in the example is the latest human protein reference from GENCODE. The protein database file will be downloaded from GENCODE website on the fly. The output folder is "pepquery_kras_g12d".
To run this command line on a computer, an internet connection is needed since it requires to access the proteomics data in PepQueryDB hosted on Amazon S3 cloud storage.
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b CPTAC_LUAD_Discovery_Study_Proteome_PDC000153 -db gencode:human -hc -o pepquery_kras_g12d/ -i LVVVGADGVGK
The above command line takes about 1 minute on a Mac Pro computer (Memory: 16 GB, Processor: 3.1 GHz Quad-Core Intel Core i7). It takes about similar time on a Linux computer (Memory: 64 GB, CPUs: 12).
The following four spectra are expected to be confidently identified in this search:
25CPTAC_LUAD_W_BI_20180901_KR_f13:26228:2 22CPTAC_LUAD_W_BI_20180820_KR_f13:27848:2 19CPTAC_LUAD_W_BI_20180730_KL_f12:26558:2 03CPTAC_LUAD_W_BI_20180521_KR_f13:25749:2
2. Search two novel peptides against a local MS/MS dataset
This example shows how to search peptides in user's MS/MS data. In this example, two novel peptides (LLSYVDDEAFIRDVAK,VGDANPALQK) are searched against an MS/MS dataset in MGF format (iPRG2015/JD_06232014_sample1-A.mgf). Both the reference database (iPRG2015/iPRG2015_no6.fasta) and the MS/MS file (iPRG2015/JD_06232014_sample1-A.mgf) used in this example can be downloaded from this link. More information about the dataset could be found in a previous publication
In this example, we consider the following parameters for MS/MS matching:
-fixMod 1 -> Fixed modification: 1 = Carbamidomethylation of C -varMod 2 -> Variable modification: 2 = Oxidation of M -tol -> Precursor mass tolerance: 10.0 ppm -itol -> Fragment ion mass tolerance: 0.05 Da Default: Enzyme: 1 = Trypsin Max Missed cleavages: 2
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -db iPRG2015/iPRG2015_no6.fasta -ms iPRG2015/JD_06232014_sample1-A.mgf -fixMod 1 -varMod 2 -tol 10 -itol 0.05 -i LLSYVDDEAFIRDVAK,VGDANPALQK -o out
The above command line takes about 30 seconds on a Mac Pro computer (Memory: 16 GB, Processor: 3.1 GHz Quad-Core Intel Core i7). It takes about similar time on a Linux computer (Memory: 64 GB, CPUs: 12).
Two spectra are identified for peptide LLSYVDDEAFIRDVAK and one spectrum is identified for peptide VGDANPALQK.
3. Search a novel peptide against multiple MS/MS datasets from PepQueryDB
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b CPTAC_LUAD_Discovery_Study_Proteome_PDC000153,CPTAC_PDA_Discovery_Study_Proteome_PDC000270,CPTAC_UCEC_Discovery_Study_Proteome_PDC000125 -db gencode:human -hc -o pepquery_kras_g12d/ -i LVVVGADGVGK
4. Search a novel peptide against all MS/MS datasets from PepQueryDB
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b all -db gencode:human -hc -o pepquery_kras_g12d/ -i LVVVGADGVGK
5. Search a novel peptide against all MS/MS datasets which contain CPTAC from PepQueryDB
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b CPTAC -db gencode:human -hc -o pepquery_kras_g12d/ -i LVVVGADGVGK
6. Search a novel peptide against all phosphoproteome datasets from PepQueryDB
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b p -db gencode:human -hc -o pepquery_kras_g12d/ -i LVVVGADGVGK
7. Search a novel peptide against all global proteome datasets from PepQueryDB
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b w -db gencode:human -hc -o pepquery_kras_g12d/ -i LVVVGADGVGK
8. Search a novel peptide against all glycosylation datasets from PepQueryDB
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b g -db gencode:human -hc -o pepquery_kras_g12d/ -i LVVVGADGVGK
9. Search a novel peptide against all acetylation datasets from PepQueryDB
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b a -db gencode:human -hc -o pepquery_kras_g12d/ -i LVVVGADGVGK
10. Search a novel peptide against all ubiquitination datasets from PepQueryDB
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b u -db gencode:human -hc -o pepquery_kras_g12d/ -i LVVVGADGVGK
11. Search multiple novel peptides against a single MS/MS dataset from PepQueryDB
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b CPTAC_TCGA_Colon_Cancer_Proteome_PDC000111 -db gencode:human -hc -o pepquery_out/ -i LVVVGADGVGK,NPLLDLAAYDQEGR
12. Search multiple novel peptides in a file against a single MS/MS dataset from PepQueryDB
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b CPTAC_TCGA_Colon_Cancer_Proteome_PDC000111 -db gencode:human -hc -o pepquery_out/ -i pep.txt
The file pep.txt looks like below:
LVVVGADGVGK
NPLLDLAAYDQEGR
13. Search a known peptide against a single MS/MS dataset from PepQueryDB
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b CPTAC_LUAD_Discovery_Study_Proteome_PDC000153 -db gencode:human -hc -o pepquery_out/ -i AHSSMVGVNLPQK -s 2
14. Validate PSMs from known peptide against a single MS/MS dataset from PepQueryDB
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b CPTAC_LUAD_Discovery_Study_Proteome_PDC000153 -db gencode:human -hc -o pepquery_out/ -i psm.txt -s 2
The file psm.txt looks like below:
AHSSMVGVNLPQK 01CPTAC_LUAD_W_BI_20180515_KR_f01:15128:4
AHSSMVGVNLPQK 01CPTAC_LUAD_W_BI_20180515_KR_f01:15349:5
15. Search a novel protein against a single MS/MS dataset from PepQueryDB
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b CPTAC_TCGA_Colon_Cancer_Proteome_PDC000111 -db gencode:human -hc -o pepquery_out/ -i MKKFFDSRREQGGSGLGSGSSGGGGSTSGLGSGYIGRVFGIGRQQVTVDEVLAEGGFAIVFLVRTSNGMKCALKRMFVNNEHDLQVCKREIQIMRDLSGHKNIVGYIDSSINNVSSGDVWEVLILMDFCRGGQVVNLMNQRLQTGFTENEVLQIFCDTCEAVARLHQCKTPIIHRDLKVENILLHDRKVFHELTQTDKMGAQELLR -t protein
16. Search a novel DNA sequence against a single MS/MS dataset from PepQueryDB
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b CPTAC_TCGA_Colon_Cancer_Proteome_PDC000111 -db gencode:human -hc -o pepquery_out/ -i GAGCCCAGGAGTTTGAGGCTGCAGTGAGTTGTGAGTCCAGCCTGGGTGACAGAGTGAGACACTGTCTCCAAAAATATAATAATAAAAATAAAGCCTGTATCTCAATGGGAAGATTTCTGCTAAGGCAGTTCAACTCACTGGAAAGAACTGCTGCATTAGGTTTCAACTTTAATGCTTTCTGTTACTTCTAGTAAAGGTTAAGTGATTTTTCCATTAGCTGTAGAAGTTTGGAGAGCTATTTACCAAGCCAGATCAATGATTTAAAAATTATTGGAAATTCATCTAAGAATCAAGTCTGAATGCCCAATTCTATTGCGGTTGATTAGGTGTGATATTCTTTAAAGTTCAGGAATATTGGCAGTAAAAAATGAGCAGCTACTTTTCAATACTTTGTCCTTTTTTGGTGGTCTCTGCCTATTTCAAATGTCCTGATCAAAAGATAAATAATTGGCACTGTGCCAAGGTTTGGTTTTCCAACTAAGGTTTCAACTGTGCCAGAAACCTATGTCCTTCACTTTGGTGGATGCTAAATGTTATTCTAAGAATATGCTTTTTCCCAATTCTCCTTTCTGATTTTTATGTATTAGTGGATGCAAAATTGTCTTTCTAGTTGAATGAATAATTTCGGCTAATGCACGTGGAACTTTGCACCCCAGATTCTTCCCATGGTCATTATCAAGTGAAGCCCTCAAAAACATGAGCGAAGAGCCTAGAAATACTCAGGGAGATTTCTCACCCCAACTCAGAAATTTTTTTTTTTTTTTTCAAGACGGCGTCTTGCTCTGTCGCTCAGGCGGGAGTGCAGTGGTGTGATCTCGGCTCACTGCAACCTCCATTTTCCGAGTTCAAGCGATTCTGCCTCAGCCTCCCGAGTAGCTGGGATTATAGGCACACACCACCGCGCCTGGCTAATTTTTGTATTCTTAGTAGAGATGGGGTTTCACCATGTTGGCCAG -t DNA
17. Search a known protein against a single MS/MS dataset from PepQueryDB
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -b CPTAC_LUAD_Discovery_Study_Proteome_PDC000153 -db swissprot:human -hc -o pepquery_out/ -i "sp|P07205|PGK2_HUMAN" -t protein -s 2
sp|P07205|PGK2_HUMAN is a protein ID in the SwissProt human protein database:
>sp|P07205|PGK2_HUMAN Phosphoglycerate kinase 2 OS=Homo sapiens OX=9606 GN=PGK2 PE=1 SV=3 MSLSKKLTLDKLDVRGKRVIMRVDFNVPMKKNQITNNQRIKASIPSIKYCLDNGAKAVVL MSHLGRPDGVPMPDKYSLAPVAVELKSLLGKDVLFLKDCVGAEVEKACANPAPGSVILLE NLRFHVEEEGKGQDPSGKKIKAEPDKIEAFRASLSKLGDVYVNDAFGTAHRAHSSMVGVN LPHKASGFLMKKELDYFAKALENPVRPFLAILGGAKVADKIQLIKNMLDKVNEMIIGGGM AYTFLKVLNNMEIGASLFDEEGAKIVKDIMAKAQKNGVRITFPVDFVTGDKFDENAQVGK ATVASGISPGWMGLDCGPESNKNHAQVVAQARLIVWNGPLGVFEWDAFAKGTKALMDEIV KATSKGCITVIGGGDTATCCAKWNTEDKVSHVSTGGGASLELLEGKILPGVEALSNM
18. Search a known gene against a proteomics dataset from a public proteomics data repository
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -varMod 2 -fixMod 0 -t protein -s 2 -ti 0,1 -tol 50 -itol 0.05 -hc -db uniprot-proteome_UP000005640_format.fasta -i F2Z2R5,F6T542,K7EMC0,K7EMY3,K7ESA5,Q8TDI0 -b "MSV000088555:j11456.mzML|b10335.mzML" -o pepquery_out/HDAC1/CHD5
F2Z2R5,F6T542,K7EMC0,K7EMY3,K7ESA5,Q8TDI0 are the protein isoforms of gene CHD5 in the protein reference database uniprot-proteome_UP000005640_format.fasta:
>Q8TDI0 Chromodomain-helicase-DNA-binding protein 5 OS=Homo sapiens OX=9606 GN=CHD5 PE=1 SV=1 MRGPVGTEEELPRLFAEEMENEDEMSEEEDGGLEAFDDFFPVEPVSLPKKKKPKKLKENK CKGKRKKKEGSNDELSENEEDLEEKSESEGSDYSPNKKKKKKLKDKKEKKAKRKKKDEDE DDNDDGCLKEPKSSGQLMAEWGLDDVDYLFSEEDYHTLTNYKAFSQFLRPLIAKKNPKIP MSKMMTVLGAKWREFSANNPFKGSSAAAAAAAVAAAVETVTISPPLAVSPPQVPQPVPIR KAKTKEGKGPGVRKKIKGSKDGKKKGKGKKTAGLKFRFGGISNKRKKGSSSEEDEREESD FDSASIHSASVRSECSAALGKKSKRRRKKKRIDDGDGYETDHQDYCEVCQQGGEIILCDT CPRAYHLVCLDPELEKAPEGKWSCPHCEKEGIQWEPKDDDDEEEEGGCEEEEDDHMEFCR VCKDGGELLCCDACPSSYHLHCLNPPLPEIPNGEWLCPRCTCPPLKGKVQRILHWRWTEP PAPFMVGLPGPDVEPSLPPPKPLEGIPEREFFVKWAGLSYWHCSWVKELQLELYHTVMYR NYQRKNDMDEPPPFDYGSGDEDGKSEKRKNKDPLYAKMEERFYRYGIKPEWMMIHRILNH SFDKKGDVHYLIKWKDLPYDQCTWEIDDIDIPYYDNLKQAYWGHRELMLGEDTRLPKRLL KKGKKLRDDKQEKPPDTPIVDPTVKFDKQPWYIDSTGGTLHPYQLEGLNWLRFSWAQGTD TILADEMGLGKTVQTIVFLYSLYKEGHSKGPYLVSAPLSTIINWEREFEMWAPDFYVVTY TGDKESRSVIRENEFSFEDNAIRSGKKVFRMKKEVQIKFHVLLTSYELITIDQAILGSIE WACLVVDEAHRLKNNQSKFFRVLNSYKIDYKLLLTGTPLQNNLEELFHLLNFLTPERFNN LEGFLEEFADISKEDQIKKLHDLLGPHMLRRLKADVFKNMPAKTELIVRVELSQMQKKYY KFILTRNFEALNSKGGGNQVSLLNIMMDLKKCCNHPYLFPVAAVEAPVLPNGSYDGSSLV KSSGKLMLLQKMLKKLRDEGHRVLIFSQMTKMLDLLEDFLEYEGYKYERIDGGITGGLRQ EAIDRFNAPGAQQFCFLLSTRAGGLGINLATADTVIIYDSDWNPHNDIQAFSRAHRIGQN KKVMIYRFVTRASVEERITQVAKRKMMLTHLVVRPGLGSKSGSMTKQELDDILKFGTEEL FKDDVEGMMSQGQRPVTPIPDVQSSKGGNLAASAKKKHGSTPPGDNKDVEDSSVIHYDDA AISKLLDRNQDATDDTELQNMNEYLSSFKVAQYVVREEDGVEEVEREIIKQEENVDPDYW EKLLRHHYEQQQEDLARNLGKGKRIRKQVNYNDASQEDQEWQDELSDNQSEYSIGSEDED EDFEERPEGQSGRRQSRRQLKSDRDKPLPPLLARVGGNIEVLGFNARQRKAFLNAIMRWG MPPQDAFNSHWLVRDLRGKSEKEFRAYVSLFMRHLCEPGADGAETFADGVPREGLSRQHV LTRIGVMSLVRKKVQEFEHVNGKYSTPDLIPEGPEGKKSGEVISSDPNTPVPASPAHLLP APLGLPDKMEAQLGYMDEKDPGAQKPRQPLEVQALPAALDRVESEDKHESPASKERAREE RPEETEKAPPSPEQLPREEVLPEKEKILDKLELSLIHSRGDSSELRPDDTKAEEKEPIET QQNGDKEEDDEGKKEDKKGKFKFMFNIADGGFTELHTLWQNEERAAVSSGKIYDIWHRRH DYWLLAGIVTHGYARWQDIQNDPRYMILNEPFKSEVHKGNYLEMKNKFLARRFKLLEQAL VIEEQLRRAAYLNMTQDPNHPAMALNARLAEVECLAESHQHLSKESLAGNKPANAVLHKV LNQLEELLSDMKADVTRLPSMLSRIPPVAARLQMSERSILSRLTNRAGDPTIQQGAFGSS QMYSNNFGPNFRGPGPGGIVNYNQMPLGPYVTDI
19. Validate a USI
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -i VLHPLEGAVVIIFK -ms "mzspec:PXD000561:Adult_Frontalcortex_bRP_Elite_85_f09:scan:17555" -db swissprot:human -hc -s 2 -o usi -itol 0.05
20. Validate a USI
java -jar pepquery-2.0.2/pepquery-2.0.2.jar -ms "mzspec:PXD010154:01284_E04_P013188_B00_N29_R1.mzML:scan:31291" -i DQNGTWEMESNENFEGYMK -itol 0.05 -hc -db swissprot:human -o usi -s 2 -varMod 2 -fixMod 1 -tol 10 -ti 0,1
Index MS/MS data for fast searching
You can index MS/MS data for fast searching by using the following command lines. This can significant speed up the search especially when the size of MS/MS data is very large.
java -cp pepquery-2.0.2/pepquery-2.0.2.jar index -h usage: Options -c The number of CPUs. Default is all available CPUs. -d Dataset ID -f File type (file suffix) to download. -h Help -i MS/MS data file or folder -m Method for Raw MS/MS data conversion: ThermoRawFileParser -o Output folder -p Tool path for Raw MS/MS data conversion: for example, /home/test/ThermoRawFileParser/ThermoRawFileParser.exe -r Delete raw file after data conversion -show Show all files without downloading any file.
Once we have the MS/MS data indexed, we can give the folder (this is the folder for parameter -o when we build MS/MS index) which containing the indexed MS/MS files to parameter -ms when we do PepQuery search. The orginal MS/MS data is not required any more for PepQuery search after we have the indexed MS/MS files.
Run PepQuery on immunopeptidomics data
PepQuery supports searching immunopeptidomics data but it may require large memory for the analysis.